Any improvement in battery performance will come about through a better understanding of the overall functioning of the various components: electrodes, electrolytes and interfaces. Two studies are presented here on positive and negative electrode materials using in operando measurements made on several SOLEIL beamlines.
Over the past twenty years, the demand for energy storage capacity has never stopped growing. Whether it is to drive an electric car, operate a mobile (phone or computer), or as solar cells or wind turbines, this always involves the production, consumption and storage of energy, with batteries as the core element.
Research is very active worldwide in this area, with the principal objective of having batteries that can store the maximum energy at the lowest cost and under optimal safety conditions.
Any improvement in battery performance will come about through a better understanding of the overall functioning of the various components: electrodes, electrolytes and interfaces. With regard to lithium batteries, two approaches are possible at the moment: the study of materials for the positive and negative electrodes. X-ray absorption spectroscopy is a characterization technique particularly well suited for exploring the electrochemical mechanisms at stake in battery electrodes, using in operando measurements (a cell specifically for in-situ measurements has been developed recently [1]). Presented here are two recent studies on positive and negative electrode materials, using measurements taken in operando on several SOLEIL beamlines.
What happens when a battery is being operated?
Recent studies have clearly shown the major role played by the formulation and synthesis of electrodes in a battery’s performance. Electrodes are composite materials, i.e. formed from a mixture of active materials (in relation to Li), a conductive agent (carbon) and a binder. The main parameters in the formulation of electrodes are particle size, the proportion of active material relative to the conductive additives and polymer binder materials, the latter providing the mechanical strength and porosity of the electrode.
The electronic percolating network and simultaneously, the diffusion of ions in the electrode are the key factors in the proper functioning of a battery.
Ideally these two charged species (electrons and lithium ions) must enter and exit particles in the same quantities and at the same speed. If this is not the case, transformation of the corresponding particle is likely to be delayed in relation to the mean state of charge determined by electrochemical measures of the whole electrode. To compensate for this delay, the well "connected" particles will be in a more advanced transformation state of lithiation or delithiation. Such behavior can result, at least before equilibrium, in significant heterogeneity in the electrode. This issue has been addressed by some research groups, either using spectroscopic methods at rest or through thermodynamic considerations. Thus, an important still pending question is: "What is the degree of homogeneity in the lithium and electron content of an electrode while in operation?”
Techniques and approaches
In order to answer this question, it is important to able to study the batteries at several time and space resolutions scales. Remember also that these electrode materials are oxidation and reduction centers that receive, during the battery’s operation, varying amounts of electrons and lithium ions. Therefore, they are continuously subject to structural and electronic changes. We used X-ray diffraction (XRD) and X-ray absorption (XAS) spectroscopy techniques known for their effectiveness in the characterization of the atomic and electronic structure of matter. Due to the high photon flux delivered by the synchrotron radiation sources and the recent technical development of new beamlines, we performed several experiments at SOLEIL (on the SAMBA, ODE, LUCIA and CRISTAL beamlines) to combine different accessible temporal and spatial resolutions.
XRD/XAS proof of an apparent delay (CRISTAL/SAMBA&ODE)
LiFePO4 is positive electrode material that has been studied at length and is currently in the marketing phase. A delay in the structural transformation of LiFePO4 into FePO4 was observed using XRD for the first time by a group of Taiwanese researchers [1]. Subsequently, the appearance of an amorphous phase was then advanced as an explanation for the delay [2]. However, our preliminary results, also obtained using XRD, have refuted the existence of an amorphous phase, since the precise fitting of the diffraction intensities collected in operando tends to prove that no amorphous phase was created, the disappearance of the LiFePO4 phase corresponding to the formation of delithiated FePO4 during two-phase charging.
Our first XAS experiments on SAMBA also confirmed this reaction delay. The presence of well-defined isobestic points, obtained on spectral evolution curves recorded at the Fe K-edge during electrode discharging and charging, is a clear sign of the two-phase nature of the system. As a result, the spectra obtained for all compositions between LiFePO4 and FePO4 are linear combinations of the spectra of the initial and final compounds. The changing proportions of the two phases, extrapolated from results of the linear combinations, confirm this apparent delay. However, the XAS technique is not sensitive to the crystalline state of the material under study and again, in our case, the formation of a third amorphous phase, in relatively large amounts, cannot simply explain this observed delay.
We therefore assumed that in the electrode some parts were "in advance" of the phase transformation, in order to meet the overall composition of the electrode given at every instant by electrochemical measurements. To probe this heterogeneity of the electrode, we simply removed, in a glove box under a controlled atmosphere, the electrode of the experimental cell after reaching the overall Li0.5FePO4 electrochemical composition. Using XAS, we then probed four distinct positions on the electrode placed in an airtight and moisture-free enclosure.
All positions considered had different spectra, which also differed from the theoretical spectrum. Two points were "delayed" and two others slightly “in advance”.
Since these spectra were collected on an electrode at thermodynamic equilibrium, we considered it important to examine the behavior of these LiFePO4 electrodes at several points during the cycle and therefore with a smaller beam. Thus, measurements were made on the ODE beamline with a spectrum obtained every 0.1s and a beam size of less than three orders of magnitude. Moving the electrode during measurements can probe simultaneously advances or delays and therefore correlated heterogeneity. Seven different points were examined while the battery was operating and again the data clearly show differences in behavior from one point to another, confirming the electrode’s heterogeneity.
An important observation to note is that, although some points were very delayed, all the points studied finally reached the overall electrochemical composition corresponding to the end of charging, proving that on this scale grains or grain assemblies (studied at these points) are connected to the electronic and ionic network, even if each is connected more or less efficiently.
Mapping of the electrode (LUCIA)
As we showed with XAS that the apparent "delay" is a consequence of the heterogeneity in the diffusion of Li+ ions or electrons, we wanted to study the electrode’s heterogeneity by mapping during operation. We conducted mapping experiments on LUCIA specially designed for such experiments with a controlled displacement of the sample and a small beam (7x7 µm2). This beamline is equipped with a standard monochromator and stepper motor that is not suitable for rapid data collection. In fact, a full XANES spectrum, such as those shown in Fig. 1, would be recorded in about 5 minutes, which is not suitable for our dynamic studies. In addition, a complete electrode map includes about 1300 points, so it would take more than 4 days to acquire a single composition, a time frame incompatible with the use of all synchrotron radiation sources.
Therefore, we conducted fixed energy experiments to map the electrodes, either during operation (3 s per point) or at different equilibrium states. This fixed energy study is possible because the process to be studied is a two-phase reaction, which implies that all energy levels, the intensity of the recorded absorption is a linear combination of the intensity of the initial and final phases, and allows direct measurement of the FeII/FeIII ratio. We chose the energy corresponding to the maximum intensity difference between initial and final states. As the absorption intensity is also correlated with the density of the element being probed (here iron) present at each point of the electrode, it is necessary to measure the density at each point before the start of the cycle then normalize the data obtained using these initial values.
The electrode was probed with a spatial resolution of 7 x 7 µm2. The selected colors were red for FeII (LiFePO4) and purple for FeIII (FePO4).
Data were collected at the same time both in the transmission mode, through a hole in the cell, and in fluorescence mode on the entire surface of the electrode. In transmission, only 12.5 mm2 of the electrode can be mapped, but the map is representative of the total thickness of the electrode. Using fluorescence, the entire surface of the positive electrode can be studied, but only to a depth of about 7 microns. Fig. 2 shows the fluorescence maps obtained in dynamic mode while charging for the extraction of a lithium ion in an hour. The distribution of the proportion of FeII is clearly heterogeneous, with certain parts of the electrode containing more LiFePO4 than expected, while other parts contain less. We also observed that the hole had a significant influence on the experimental cell (dashed lines). This is probably due to the pressure difference applied to the position of the hole, where the electrical contact with the current collector is less effective. In this case, heterogeneity is attributed to limited electron transfer. This important observation provides a reasonable explanation for the apparent delay in transmission observed by several authors using XRD. The area exposed for XRD shows a "delay" caused by a smaller electrical contact (lower pressure) at the center of the electrode.
The strong influence of regime on the heterogeneity of the electrode was also highlighted through such maps. More information on this can be found in the article (G. Ouvrard, M. Zerrouki, P. Soudan, B. Lestriez, C. Masquelier, M. Morcrette, S. Hamelet, S. Belin, A. M. Flank, F. Baudelet J. of Power Sources 229 (2013) 16-21).
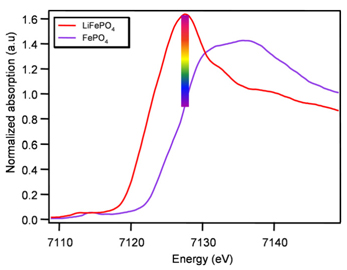
Fig1 X-ray absorption spectra of LiFePO4 (red), FePO4 (purple). Maximum of absorption difference between these two phases is indicated, and colour code attributed for the mean iron oxidation state.
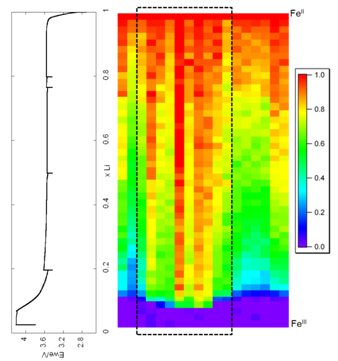
Fig. 2 Normalized dynamic maps recorded at 7127 eV in fluorescence mode during the charge of the battery.
Références :
[1] J.B. Leriche, S. Hamelet, J. Shu, M. Morcrette, C. Masquelier, G. Ouvrard, M. Zerrouki, P. Soudan, S. Belin, E. Elkaïm, F. Baudelet, J. Electrochem. Soc. 157 (2010) A606-A610.
[2] H.H. Chang, C.C. Chang, H.C. Wu, M.H. Yang, H.S. Sheu, N.L. Wu, Electrochem. Comm. 10 (2008) 335-339.
[3] H.C. Shin, K.Y. Chung, W.S. Min, D.J. Byun, H. Jang, B.W. Cho, Electrochem. Comm. 10 (2008) 536-540.
Mechanism of NbSnSb a new negative electrode materials for Li-ion batteries
Recently tested as negative electrode in Li-ion batteries, TiSnSb has showed to be extremely promising electrode material by sustaining a capacity over 500 mAh/g for more than 250 cycles, even at fast cycling rates (4C) and with an excellent 99 % coulombic efficiency. From operando XRD and Mössbauer spectroscopy measurements [1, 2], a conversion mechanism has been observed leading simultaneously to the formation of Li3Sb and Li7Sn2 alloys, but no information was obtained about Ti. Upon charge, a re-conversion has been demonstrated leading to an alloy structurally close to the pristine material but still undefined. Moreover the good electrochemical performance of TiSnSb (compared to Sn, Sb or SnSb) would be correlated to the presence of the non-active metal (vs Li) i.e. Ti. Among the rare ternary phases in the system M/Sn/Sb (M = transition metal) only NbSnSb presents both the same stoichiometry than TiSnSb and a close structure. We decided to explore the whole system Ti1-yNbySnSb with 0 ≤ y ≤ 1 to better understand the role played by the transition metal on both the mechanism and the performance of the battery. XANES and EXAFS of the whole series of Ti1-yNbySnSb were performed before and during the cycles of the battery (with a specific designed cell elaborated during the PULSSE ANR project).
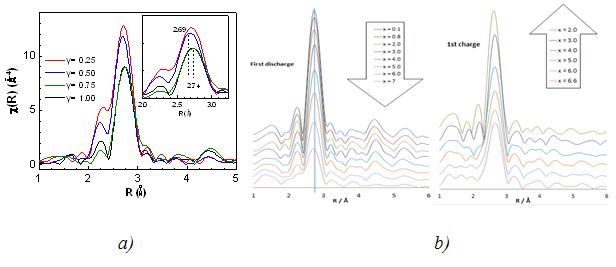
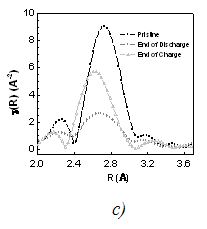
FT of EXAFS for Ti1-yNbySnSb (a), during 1st discharge/charge of the NbSnSb/Li (b) and for selected spectra: pristine, after discharge and after charge (c)
The normalized K-edge spectra for all Ti1-yNbySnSb samples were quite identical meaning that Nb remains roughly in the same electronic state. The pseudoradial distribution derived from EXAFS signals (Fig 1a) shows that the main peak is centred around 2.69 Å for Ti1-yNbySnSb with y = 0.25 and 0.5 and around 2.74 Å for Ti1-yNbySnSb with y = 0.75 and 1. This trend is consistent with the M-M and the M-Sn/Sb distances slightly shorter for M = Ti than for M = Nb if and only if we consider the existence of a single phase made up of tetragonal (NbSnSb type) and orthorhombic (TiSnSb type) crystallographic domains. Note that XRD was not able to give clear information about the phase composition of the different Ti1-yNbySnSb samples. The Nb K edge is quite unchanged during the discharge/charge of the NbSnSb based electrode (Fig 1b). From the analysis of the pseudoradial distribution function (Fig 1c) during the cycling of the battery NbSnSb/Li we analyzed that the broad peak (in black) centered at 2.73 Å which corresponds to the Nb-Nb, Nb-Sn and Nb-Sb bonds in the starting NbSnSb. It disappears to give a broad peak centered at 2.68 Å that could correspond to the Nb-Nb bond in cubic metallic niobium phase. The best fit was obtained by adding a Sb (or a Sn) atom in the first coordination sphere of Nb in the cubic niobium cell. Furthermore the phase diagram of Nb/Sb exhibits a phase with 4%atom of Sb inclusion in the Nb structure. After the charge, the broad peak centered at 2.68 Å is difficult to fit.
The EXAFS analysis has helped to demonstrate that Ti1-yNbySnSb are single phases made of TiSnSb-NbSnSb crystallographic domains in various amount function of the Ti/Nb composition. From operando recorded spectra we demonstrated the reversibility of the electrochemical mechanism. Moreover for the first time in the study of conversion type material, it was demonstrated that a small amount of Sb (and/or Sn) from the Li3Sb (or LixSn) matrix diffuses in the nanosized transition metal particles (here Nb) in the fully discharged electrode, demonstrating the strong metastability of the electrode in that discharged state.
References:
[1] M.T. Sougrati, J. Fullenwarth, C. Marino, B. Fraisse, J.C. Jumas, L. Monconduit J. Mater. Chem., 2011, 21 (27), 10069 - 10076
[2] H. Wilhelm, C. Marino A. Darwiche, L. Monconduit, B. Lestriez Electrochemistry Communications 24, 2012, 89–92