Le couplage des mouvements électroniques et nucléaires est une manifestation frappante de la dynamique complexe pluri-particules mise en jeu dans les systèmes hors équilibre tels que les molécules excitées.
Dans le cas des transitions électroniques moléculaires, un tel couplage peut être révélé par la rupture de l'approximation dite de Franck-Condon (FC). Cette approximation est largement acceptée, tout comme son hypothèse de transitions verticales dans l'approximation de Born-Oppenheimer (séparation des mouvements des noyaux et des électrons justifiée par leurs masses très différentes). De fait, l’approximation de FC consiste à considérer que la probabilité d'une transition électronique donnée ne dépend pas de la géométrie nucléaire échantillonnée pendant le mouvement vibrationnel. Par conséquent, on peut s'attendre à une dynamique électrons/noyaux entièrement découplée. Dans le cas d'une photoionisation moléculaire, cela signifie que les propriétés du continuum électronique doivent être découplées des spectres d'énergie vibrationnelle de l'ion résultant. Des écarts à ce modèle peuvent bien sûr être rencontrés, et sont le plus souvent attribuables aux résonances de forme dans le continuum.
Dans ce contexte, les systèmes moléculaires chiraux peuvent offrir des possibilités uniques d'étudier la photoionisation moléculaire (dont la dynamique vibrationnelle) de manière très détaillée, même pour les échantillons orientés au hasard. En effet, pour de tels systèmes chiraux, la distribution angulaire des photoélectrons dans le référentiel du laboratoire produite par photoionisation en lumière polarisée circulairement (LPC) peut présenter une forte asymétrie avant-arrière par rapport à l'axe de la lumière, un effet nommé Dichroïsme Circulaire de Photoélectrons (PECD). Cet effet peut survivre à une moyenne sur des orientations moléculaires aléatoires, car la LPC définit une direction de l'espace par rapport à laquelle les différents atomes d'un énantiomère donné seront toujours vus dans la même configuration par le photon incident.
Quantitativement, l'asymétrie induite par le PECD est égale à 2b1, où b1 est le paramètre dit dichroïque qui inclut la contribution chirale et est antisymétrique par échange de l'énantiomère ou de l’hélicité de la lumière. Il a été montré que les mesures PECD peuvent être utilisées pour sonder la géométrie et les structures (statiques) moléculaires, dont les conformères et les substitutions chimiques. Le PECD apparaît donc aussi comme un outil idéal pour étudier l'interaction dynamique entre les mouvements électroniques et nucléaires suite à la photoionisation.
Le cas du méthyloxirane (fig.2), photoionisé juste au-dessus de son énergie d'ionisation (une région située loin de toute résonance de forme) avec la lumière polarisée circulairement VUV de la ligne de lumière DESIRS, a été étudié. Les électrons correspondants ont ensuite été imagés sur un détecteur sensible en position par un spectromètre d'imagerie de vitesse (VMI) qui permet d'effectuer une spectroscopie de photoélectrons résolue en angle (AR-PES) multiplexée avec une efficacité de collection de 100 % et une résolution en énergie cinétique de 5 %.
La distribution radiale des images de photoélectrons permet d'obtenir le PES, tandis que les images de différence LPCdroite-LPCgauche fournissent le paramètre dichroïque b1. Les images de différence (Fig.1 a et b) révèlent une structure vibrationnelle nette dans la distribution radiale (correspondant à l'énergie des électrons), associée à de fortes asymétries avant-arrière dans la distribution angulaire. Des changements de signe brutaux de cette dernière quantité pour certains niveaux vibrationnels adjacents sont aussi visibles dans la cartographie (avec alternance des couleurs).
La structure vibrationnelle identifiée dans le PES (Fig.1c), c'est-à-dire la bande origine (pic « a ») et une série d'excitations uniques vibrationnelles -des déformations du squelette- (pics « b » à « f »), présentent un comportement dichroïque très caractéristique et remarquable : des valeurs b1 (Fig.1d) passant d'une valeur nulle pour le pic « a » à une valeur négative de -0,03 pour « c » puis changeant de signe quelques dizaines de meV plus loin pour atteindre ~ +0,02 pour les pics « d » et « e ». D'un point de vue phénoménologique, ce changement de signe de b1 signifie que l'asymétrie avant/arrière change de direction pour des modes vibrationnels adjacents résolus individuellement. À notre connaissance, ce comportement surprenant de basculement de l'asymétrie suite à une excitation vibrationnelle (adjacente) n'avait été observé dans aucune expérience de photoionisation moléculaire. D'autre part, cette dépendance du PECD par rapport au spectre d'énergie vibrationnelle du cation résiduel est complètement inattendue dans le cadre des hypothèses de FC.
Cet effet vibrationnel frappant est probablement dû à la sensibilité notoire du PECD par rapport à la structure moléculaire statique, qui serait d'une certaine manière répliquée dans les changements structuraux dynamiques causés par le mouvement vibrationnel, échantillonnant différentes régions de l'espace des configurations nucléaires.
Cet effet vibrationnel de photoionisation en PECD apparaît comme plus remarquable que les effets vibrationnels mesurés par le passé sur des molécules fixées dans l'espace. Toutefois, il a été obtenu sans nécessiter une orientation moléculaire particulière en raison de la nature intrinsèquement chirale des cibles. Dans ce contexte, le PECD apparaît comme une sonde puissante et universellement applicable (du moins pour les espèces chirales) de la dynamique vibrationnelle dans la photoionisation moléculaire, même pour des espèces orientées au hasard. Ceci correspond bien sûr à la situation « normale » pour les molécules chirales, qui sont omniprésentes dans la biosphère.
Outre son intérêt fondamental, cette découverte pourrait avoir des conséquences importantes pour l'interprétation du PECD dans un contexte analytique, notamment dans le domaine en expansion rapide des expériences laser de PECD à plusieurs photons, dont les résultats peuvent être influencés par la dynamique vibrationelle tant dans l'état intermédiaire que dans l'état final.
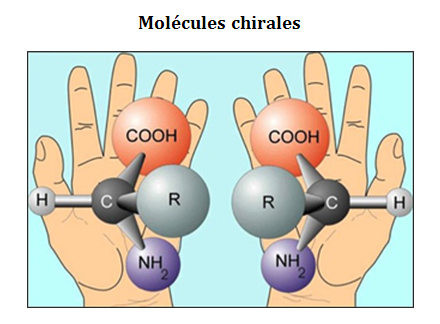
Les espèces chirales (tout comme les mains) existent sous deux formes, dites énantiomères droit et gauche, qui ne peuvent être superposées mais sont des images-miroir l'une de l'autre. Ces deux énantiomères ont exactement les mêmes propriétés physiques et chimiques, excepté lorsqu'ils sont incorporés dans un milieu lui-même chiral, ce qui conduit alors à des processus énantio-sélectifs et au phénomène dit de reconnaissance chirale (par lequel un pied droit peut par exemple reconnaître une chaussure droite !). En chimie, un tel milieu chiral peut être constitué d'autres espèces chirales formant des complexes chiraux. En physique, ce milieu chiral peut être de la lumière polarisée circulairement : pour une hélicité donnée, cet objet chiral interagit différemment avec les énantiomères gauche et droit, un effet chiroptique nommé dichroïsme circulaire (DC).
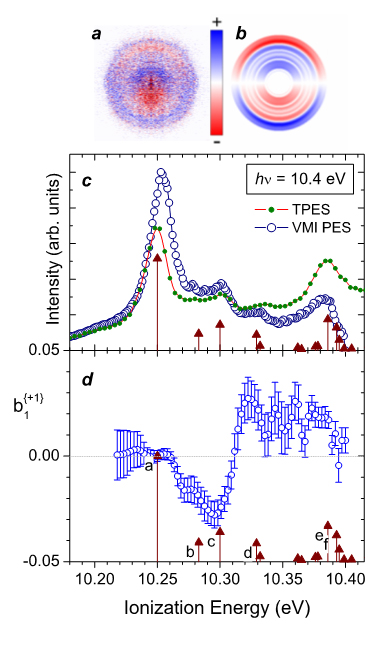
PES et paramètre dichroïque à 10,4 eV pour l'orbitale HOMO du S-méthyloxirane. Images-différence (LPCGauche – LPCDroite) filtrées sur la masse de l'ion parent: (a) brute et (b) traités par inversion d'Abel obtenues sur le spectromètre VMI. À noter que le faisceau de photons se propage de bas en haut sur cette image. (c) VMI-PES. (d) Courbe du paramètre dichroïque . Les flèches correspondent au spectre vibrationnel calculé.